INTRODUCTION
The neuropathology of the human prion diseases (Creutzfeldt-Jakob disease (CJD) Gerstmann-Straussler-Scheinker syndrome (GSS) and kuru is characterised by 4 features: spongiform change, neuronal loss, astrocytosis and amyloid plaque formation. These features are shared with prion diseases in animals, and the recognition of these similarities prompted the first attempts to transmit a human prion disease (kuru) to a primate in 1966, followed by CJD in 1968 and GSS in 1981. These neuropathological features have formed the basis of the histological diagnosis of human prion diseases for many years, although it was recognised that these changes are enormously variable both from case to case and within the central nervous system (CNS) in individual cases. It is interesting to note that the original case reported by Creutzfeldt and 2 of the original cases reported by Jakob do not show any of these characteristic neuropathological features; the diagnosis in these cases remains uncertain upon review. However, at least 2 of Jakob's original cases show typical neuropathological changes and other cases subsequently reported from his laboratory (including the members of the Backer family) also exhibited classical histological features. It is also of interest to note that prion protein (PrP) gene analysis has recently been performed on one of the Backer family cases, showing a codon 178 Asn mutation with met/val at codon 129. This genotype has been described in other familial forms of human prion disease.
Early neuropathological reports on human prion diseases suffered from a confusion of nomenclature, in which the significance of the diagnostic feature of spongiform change was occasionally overlooked. The subsequent demonstration that human prion diseases were transmissible reinforced the importance of spongiform change as a diagnostic feature, reflected in the use of the term `spongiform encephalopathy' for this group of disorders. Recent advances in the understanding of the infectious agent, along with increasing knowledge on the pathogenetic significance of mutations and polymorphisms in the human prion protein gene have prompted a revaluation of classical neuropathology in this group of diseases, and a tendency to use the generic term `prion disease' rather than spongiform encephalopathy.
Neuropathological assessment of the structural changes in the CNS has been the mainstay in diagnosis of human prion diseases for many years. A new range of investigative techniques, including PrP gene analysis, PrP immunocytochemistry and detection by the Western blot, histoblot and immunoblot techniques, prion rod/SAF detection by electronmicroscopy and transmissibility to both wild-type and transgenic laboratory animals all now have diagnostic applications. In the laboratory investigation of human prion diseases a combined morphological, immunocytochemical and molecular genetic approach is desirable. However, many cases can be diagnosed on morphological assessment alone, including the vast majority of cases of sporadic CJD.
HISTOLOGICAL INVESTIGATION
Most neuropathological studies in human prion diseases are performed on paraffin-embedded tissues. Tissue blocks from the CNS can be decontaminated in 96% formic acid for 1 hour prior to processing into paraffin wax. Sections are then cut for microscopy and stained for routine analysis with haematoxylin and eosin. Spongiform change is most easily recognised at this tissue section thickness; the use of thick ( 10 microns or more) sections carries the danger of misinterpretation of other sponge-like changes in the cerebral cortex ( see below).
In most cases of human prion diseases the histological
features are distinctive and will allow a diagnosis to be reached without
undue difficulty. In CJD, the most consistent histological abnormality
is spongiform change, which is characterised by a fine vacuole-like appearance
in the neuropil, with vacuoles varying from 20-200 microns in diameter
(Figure
1). These vacuoles can appear in any layer
of the cerebral cortex and may become confluent, resulting in large vacuoles
which substantially distort the cortical cytoarchitecture. Vacuolation
may also be seen within the cytoplasm of larger neurones within the cortex.
Cortical involvement is detectable in most cases of CJD, and is usually
accompanied by spongiform change in the basal ganglia, thalamus and cerebellar
cortex. Cerebellar involvement is present in most cases, although the severity
and distribution of the spongiform change is markedly variable. Confluent
spongiform change is unusual in the cerebellum, which may however exhibit
a widespread microvacuolar change with smaller vacuoles 20-50 microns in
diameter in the molecular layer (Figure
2).

Figure 1.
Spongiform change in CJD consists of numerous rounded vacuoles within the
neuropil which occur both singly and in confluent groups, distorting the
cortical cytoarchitecture.
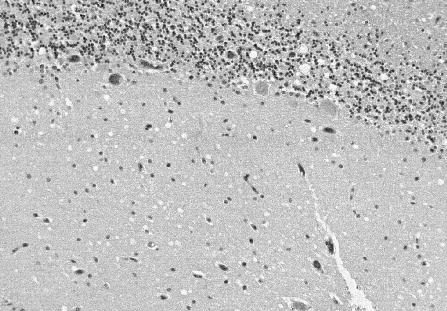
Figure 2.
Spongiform change in the cerebellum comprises multiple small vacuoles in
the molecular layer which usually do not appear confluent.
In long-standing cases, the neuronal loss and
spongiform change maybe so severe as to result in status spongiosus, where
widespread coarse vacuolation results in collapse of the cortical cytoarchitecture,
leaving an irregular distorted rim of gliotic tissue containing few remaining
neurones. The basal ganglia and thalamus may also exhibit severe neuronal
loss with gliosis and atrophy, and in the cerebellum there is often an
irregular loss of neurones in the granular cell and Purkinje cell populations.
Spongiform change in most brain regions is accompanied by neuronal loss
and gliosis involving both astrocytes and microglia (Figure 3). Microglial
hypertrophy and hyperplasia occur in a widespread distribution within the
CNS in CJD, and microglia are also implicated in the pathogenesis of PrP
plaques (see below).
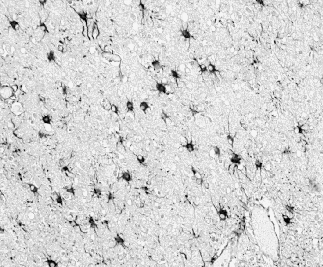
Figure 3. Astrocytosis
is widespread in CJD, with increased numbers of enlarged astrocytes in
the cerebral cortex. This section of tissue has been stained immunocytochemically
for Glial Fibrilliary Acidic Protein.
A range of clinical and neuropathological variants in sporadic CJD have been described, the most striking of which (the panencephalic variant) contained extensive necrotising lesions in the white matter. The histological features in these rare variants have largely been decribed on the basis of traditional neuropathological staining techniques; further immunocytochemical studies for PrP are required in order to more fully characterise these unusual findings.
Around 10% of CJD cases contain PrP plaques which
are usually visible as rounded eosinophilic structures. These are most
frequently observed in the cerebellum, where they usually occur with a
hyaline eosinophilic core and a paler halo (Figures 4 & 5). In kuru,
these plaques often showed a peripheral margin of radiating fibrils, and
similar changes can identified within plaques in sporadic and familial
CJD. Plaques are identified outwith the cerebellum in a minority of these
cases in the thalamus, basal ganglia or cerebral cortex. The occurrence
of plaques is related to PrP genotype and is associated with codon 129
polymorphism (being commoner in met/val or val/val genotypes); PrP plaques
have also been reported in familial CJD in association with several different
PrP gene mutations. Ultrastructural and immunocytochemical studies in both
human and animal prion diseases have demonstrated that microglial cells
are intimately involved in PrP plaque formation, and may perhaps play a
role in the processing of PrP into an amyloid structure.

Figure 4. Cerebellar
amyloid plaques in sporadic CJD consist of a highline core surrounded by
a pale halo.

Figure 5.
Prion protein immunostaining showing amyloid plaques.
OTHER HISTOLOGICAL FEATURES IN HUMAN PRION DISEASES
A range of other histological abnormalities have been described in human prion diseases, some of which relate to the affect of ageing on the human brain and have no specific association with this group of disorders. These abnormalities include swollen cortical neurones and amyloid angiopathy; other changes are summarised in Table 1. It is critically important to be aware of age-associated histological abnormalities in the human brain and not to interpret these as being indicative of a coexisting CNS disorder. Alzheimer's disease and CJD been described concurrently, although this appears to be an exceptional event. Other more apparently specific abnormalities have been described in cases of CJD which have been studied by ubiquitin immunocytochemistry. These abnormalities include dot-like ubiquitinated strutures in the neuropil, within neurones and around PrP plaques. The latter probably represent dystrophic neurites, while the intracellular lesions may represent lysosomal structures, as suggested by animal scrapie models.
Table 1 Histological changes in human prion diseases
Classical changes spongiform change neuronal loss astrocytosis PrP amyloid plaques Other changes status spongiosus neuronal swelling abnormal neuritic dendrites white matter necrosis and cavitation microgliosis beta protein amyloid angiopathyOther Conditions Associated with Spongiform-Like Change in the CNS
The importance of spongiform change as one of the histological hallmarks of human prion diseases is widely accepted, and clear distinctions have been made between spongiform change and status spongiosus. Status spongiosus can result from any neurodegenerative disorder which results in widespread neuronal death and collapse of the cerebral cortical cytoarchitecture. It is commonly encountered in Pick's disease and may also occur in Alzheimer's disease, cortical ischaemia and as a consequence of viral encephalitis. Spongiform change per se is not pathonomonic for human prion diseases. Appearances identical to spongiform change have been described in other neurodegenerative disorders (see Table 2), particularly in Alzheimer's disease and diffuse Lewy body disease. In these disorders spongiform change is usually confined to layer 2 of the cerebral cortex and is present in a restricted distribution in the frontal lobes, cingulate gyrus, temporal poles and inferior temporal cortex. In occasional cases of Alzheimer's disease and diffuse Lewy body disease the spongiform change may be particularly conspicuous and necessitate widespread histological sampling along with additional investigative techniques, including PrP immunocytochemistry, to investigate the possibility of human prion disease. It should also be recalled that other neurodegenerative disorders resulting in focal spongiform-like change are associated with distinctive lesions which help clarify diagnosis. Other disorders which may cause a sponge-like appearance in the CNS are listed in Table 2. In these disorders, confusion with spongiform change in human prion disease is not a major difficulty because of the clinical and pathological context in which these changes occur.
Table 2 CNS disorders with focal spongiform change
Alzheimer's disease Pick's disease diffuse Lewy body disease dementia of frontal lobe type dementia in motor neurone disease Sponge - like changes in other CNS disorders Grey matter status spongiosus oedema metabolic encephalopathies neuronal storage disorders tissue fixation and processing artefacts White matter oedema ischaemia metabolic encephalopathies spongy degeneration of the white matter in infancy Canavan's disease tissue fixation and processing artefacts
