Prions are small proteinaceous infectious particles which are capable
of causing several degenerative disorders of the central nervous system.
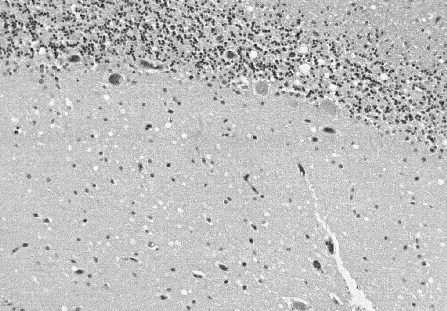
Prion diseases are often described as spongiform encephalopathies due to
the post mortem appearance of large vacuoles in the cortex and cerebellum
of the brain. In recent years, several animal and human diseases
have been identified as prion diseases, including bovine
spongiform encephalopathy (BSE or Mad Cow
Disease), scrapie (among
sheep), Kuru
(among
the Fore people of Papua New Guinea), and Creutzfeld-Jacob
Disease
(in humans).
Each of these diseases is characterized by
a slow degeneration of the central nervous system leading inevitably to
death.
Prions are small proteinaceous infectious particles which are capable
of causing several degenerative disorders of the central nervous system.
Prion diseases are often described as spongiform encephalopathies due to
the post mortem appearance of large vacuoles in the cortex and cerebellum
of the brain. In recent years, several animal and human diseases
have been identified as prion diseases, including bovine
spongiform encephalopathy (BSE or Mad Cow
Disease), scrapie (among
sheep), Kuru
(among
the Fore people of Papua New Guinea), and Creutzfeld-Jacob
Disease
(in humans).
Each of these diseases is characterized by
a slow degeneration of the central nervous system leading inevitably to
death.
The most common prion disease is scrapie,
found in sheep and goats. Victims of scrapie tend to become irritable
and develop an intense itch, causing them to scrape off their wool or hair
(leading to the term "scrapie"). As a result of neurodegeneration,
sheep infected with scrapie also exhibit a gradual loss of muscular coordination
to the point they can no longer stand. A closely related disease
is BSE or Mad Cow Disease, in which cows become increasingly uncoordinated
and apprehensive. Post mortem examination of afflicted bovine reveals
large holes in the brain, similar to those found in sheep afflicted with
scrapie. An outbreak of BSE occurred in Great Britain in the late
1980s, which was linked to a food supplement that included meat and bone
meal from dead sheep. As a result, the British government banned
the use of animal-derived feed supplements to prevent any further outbreaks.
In humans, several prion diseases have been positively identified, including
Kuru,
Creutzfeld-Jacob
Disease (CJD), Gerstmann-Straussler-Scheinker
Syndrome(GSC), Fatal
Familial Insomnia(FFI) and Alpers
Syndrome. Each is characterized by gradual
loss of motor control, dementia, paralysis, and eventual death (typically
following pneumonia). In post mortem examinations, people afflicted
with prion diseases exhibit non-inflammatory lesions, vacuoles in brain
tissue, amyloid protein deposits and astrogliosis. Unlike viral or
bacterial infections, people afflicted with prion diseases exhibit any
sign of inflammation or fever, indicating the immune system is not activated
by prion infection. Each of these ailments has similar symtoms to
animal prion diseases and each other, particularly the spongiform appearance
of the brain, suggesting they are "closely related".
Kuru
is
the most isolated human prion disease, limited to the Fore people of Papua
New Guinea. It was first described in 1957 by Vincent Zigas and D.
Carleton Gaidusek who noted that many people in this geographically isolated
tribe became afflicted by a fatal disease marked by the loss of muscular
coordinaion and dementia. Kuru has a slow onset period and is characterized
by cerbral difficulties, shaking ataxia, congestion of the blood vessels
and cortical atrophy, leading to death within 18-24 months. The cause
of the strange disease was traced to ritual cannibalism within the tribe
(the Fore people honored their dead by eating a soupprepared from the deceased's
brain). Since then, the practice has ceased and kuru has rapidly
declined, almost to the point of extinction. Interestingly, none
of the other tribes in the region who practice the same ritual are afflicted
with kuru, leading researchers to believe that at some point in the tribe's
history, one person developed a prion disease which was passed on to the
tribe when the afflicted person's brain was consumed.
A similar disease is Creutzfeld-Jacob
Disease(CJD), which occurs worldwide in
people around age 60. Approximately 1 per million people become afflicted
with CJD, although an estimated 1 in 10,000 people are infected at the
time of death (thesenumbers are approximations because CJD maybe confused
with other neurological disorders). Approximately 10 to 15 percent
of the cases result from inheritance, while a small number of cases occur
via intragonic infection (ie, the disease is passed on directly through
corneal transplantation, contaminated surgical instruments, and injection
of growth hormone derived from human pituitaries). The remaining
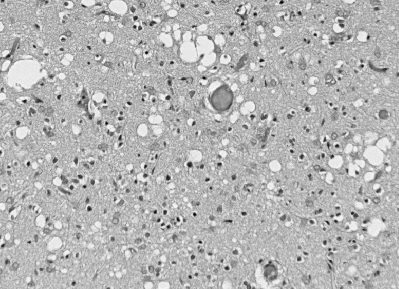
cases occur from sporadic infection. The most consistent sign of
CJD is the spongiform appearance of the brain due to vacuoles arising from
the neuronal cytoplasm which range from 20 to 200 microns in diameter.
This photograph shows the spongiform appearance of brain of a CJD victim.
Note the large vacuoles which occur both singly and in groups. |